Deux hémoglobinopathies, un traitement ingénieux couronné par le Prix Galien
La drépanocytose sévère et la ß-thalassémie dépendante des transfusions sont aujourd’hui traitables par une technologie ingénieuse d’édition de gènes ex vivo utilisant pour la première fois des "ciseaux moléculaires" sur les cellules souches hématopoïétiques du patient. Le résultat est l’exa-cel du laboratoire VERTEX, récompensé par le dernier Prix Galien de l’innovation technologique.
 Cette découverte fournit l’opportunité de remettre en lumière ces deux maladies rares, et le chemin parcouru pour soigner de mieux en mieux ces patients. Une success story racontée par les Prs Colard (HUB Hôpital Erasme), Algeri (Italie) et Essa (Arabie Saoudite) lors du 40e congrès de la Belgian Haematology Society.
Cette découverte fournit l’opportunité de remettre en lumière ces deux maladies rares, et le chemin parcouru pour soigner de mieux en mieux ces patients. Une success story racontée par les Prs Colard (HUB Hôpital Erasme), Algeri (Italie) et Essa (Arabie Saoudite) lors du 40e congrès de la Belgian Haematology Society.
L’épidémiologie
Environ 5,2 % de la population mondiale sont affectés par une hémoglobinopathie. Les estimations récentes font état de dix millions de personnes atteintes de drépanocytose (Sickle Cell Disease ou SCD) et 50.000 nouveau-nés par an atteints de bêta-thalassémies transfusions-dépendantes (TDT). La plupart des cas de TDT se retrouvent dans le bassin méditerranéen, au Moyen-Orient et en Asie, et les SCD surtout en Afrique et en Inde.
Pour le Pr Colard, « ce sont des pathologies sévères qu’on a tendance à sous-estimer, à ne pas dépister, alors que le paysage épidémiologique est en train de changer avec les migrations de populations et des explosions démographiques dans certains pays comme le Nigeria. La Belgique était un peu à la traîne jusqu’en 2023 en ne pratiquant pas le dépistage systématique des nouveau-nés. Aujourd’hui il est pratiqué, mais seulement en Fédération Wallonie-Bruxelles. »
La clinique des SCD/ TDT
Les manifestations cliniques des SCD sont une anémie, des douleurs intenses dues aux crises vaso-occlusives (CVO) et les atteintes chroniques d’organes qui affectent la qualité de vie. L’espérance de vie est réduite, avec un âge moyen de décès en Europe d’environ 40 ans. Les transfusions (à vie) sont difficiles, notamment en raison d’un taux élevé d’allo-immunisations érythrocytaires dues à la maladie elle-même et à la discordance de groupes sanguins entre les donneurs européens et les patients afrodescendants. Le traitement comporte l’hydroxyurée qui augmente le taux d’Hb fœtale, réduit la falciformation des globules rouges, prévenant ainsi les CVO douloureuses.
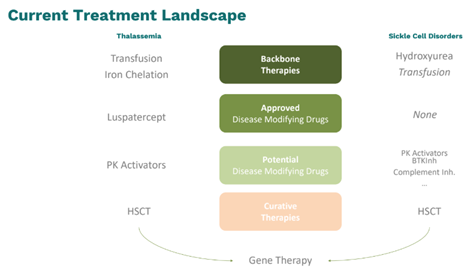 Dans les TDT, les patients présentent des anémies chroniques souvent sévères avec fatigue et essoufflements, une hématopoïèse extramédullaire requérant des transfusions à vie couplées à une chélation du fer pour éviter la surcharge ferrique et les atteintes d’organes. La qualité de vie est aussi affectée et l’âge moyen des décès en Europe se situe entre 50 ans et 55 ans. Les options thérapeutiques sont l’hydroxyurée (off label) et le luspatercept qui va améliorer l'érythropoïèse. Parmi les produits en développement, les plus prometteurs sont les activateurs de la pyruvate kinase (mitapivat, étavopivat). Pour les deux hémoglobinopathies, l’allogreffe de cellules souches hématopoïétiques (CSH) est le seul traitement à visée curative.
Dans les TDT, les patients présentent des anémies chroniques souvent sévères avec fatigue et essoufflements, une hématopoïèse extramédullaire requérant des transfusions à vie couplées à une chélation du fer pour éviter la surcharge ferrique et les atteintes d’organes. La qualité de vie est aussi affectée et l’âge moyen des décès en Europe se situe entre 50 ans et 55 ans. Les options thérapeutiques sont l’hydroxyurée (off label) et le luspatercept qui va améliorer l'érythropoïèse. Parmi les produits en développement, les plus prometteurs sont les activateurs de la pyruvate kinase (mitapivat, étavopivat). Pour les deux hémoglobinopathies, l’allogreffe de cellules souches hématopoïétiques (CSH) est le seul traitement à visée curative.
Pourquoi ne pas se contenter des allogreffes ?
L'allogreffe de CSH donne d’excellents résultats, autant dans les thalassémies que dans les drépanocytoses, avec des taux de succès proches de 90 %, variables selon les pays, les donneurs mais aussi l'âge des patients, leurs comorbidités, etc. Mais il y a le revers de la médaille.
Pour le Pr Colard, « la survie est globalement bonne, mais il ne faut pas oublier les risques liés à la procédure, l’aplasie médullaire et la réaction du greffon contre l’hôte (Graft Versus Host Disease) observée dans environ 20 % des cas en aigu, 14 % en chronique. Le taux de mortalité lié à la greffe est de 6 %, ce qui n’est pas négligeable alors qu’on dispose de médicaments efficaces qui allongent la survie. Les meilleurs donneurs (frères ou sœurs HLA compatibles) ne sont pas légion, et les taux de succès chutent au-delà de 15 ans, ce qui est l’âge de la majorité de nos greffés. »
Ce qui revient à prendre le risque de contracter une maladie pour en guérir une autre. Dans ce contexte, il faut trouver de nouveaux traitements comme les activateurs de pyruvate kinase ou les inhibiteurs de la Bruton tyrosine kinase, ou se tourner vers la thérapie génique qui est une autogreffe de CSH modifiées avec notamment, un outil d’édition génique CRISPR-Cas9.
Les thérapies géniques, du laboratoire au lit du patient
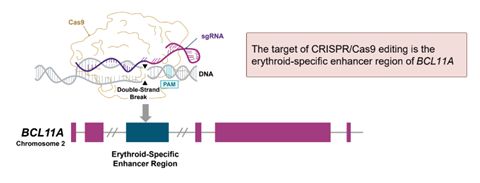 L’édition génomique permet d’effectuer des modifications génétiques ciblées dans tout type de cellule, grâce à des nucléases ou ciseaux moléculaires spécifiques comme la technologie CRISPR-Cas9. Le principe est d’isoler les CSH CD34+ du patient, les modifier à l’aide de l’outil CRISPR-Cas9 pour y désactiver le gène BCL11A afin de réactiver la production d’hémoglobine fœtale (HbF) et ensuite les réinjecter au patient.
L’édition génomique permet d’effectuer des modifications génétiques ciblées dans tout type de cellule, grâce à des nucléases ou ciseaux moléculaires spécifiques comme la technologie CRISPR-Cas9. Le principe est d’isoler les CSH CD34+ du patient, les modifier à l’aide de l’outil CRISPR-Cas9 pour y désactiver le gène BCL11A afin de réactiver la production d’hémoglobine fœtale (HbF) et ensuite les réinjecter au patient.
Le résultat est l’exa-cel (exagamglogeneautotemcel), qui n’utilise pas un vecteur viral comme dans d’autres formes de thérapie génique. Le bénéfice est impressionnant, avec 94,5 % des patients TDT (12-35 ans) qui se passent de transfusions avec exa-cel pour une durée moyenne de 40,5 mois (suivi médian de 44,7 mois) dans l’étude CLIMB THAL-111 [1], et 91,1 % des patients SCD qui ne font plus de CVO avec exa-cel durant en moyenne 35 mois (suivi médian de 39,8 mois) dans l’étude CLIMB SCD-121 [2].
Le profil de sécurité est compatible avec le conditionnement myéloablatif et une autogreffe de CSH. Les résultats sont encore meilleurs chez des patients TDT pédiatriques (2-11 ans), avec 100 % sans transfusions à 12 mois dans l’étude CLIMB THAL-141 [3], et 100 % sans CVO à 12 mois dans l’étude CLIMBSCD-151 [3]. Exa-cel améliore aussi la qualité de vie des patients TDT et SCD sur base du score HRQoL.
Et en vraie vie ? La réponse nous vient de l’Arabie Saoudite, un pays qui a des taux de prévalence de TDT et de SCD élevés par rapport aux pays voisins du Moyen-Orient et une population très homogène. Des algorithmes décisionnels ont été développés pour sélectionner les patients, et les protocoles sont très standardisés à toutes les étapes depuis le recrutement des patients, le conditionnement, la préparation de la greffe, sa manipulation jusqu’au suivi après sortie de l’hôpital. Toutes les données démographiques, cliniques, biologiques et techniques sont encodées dans un registre, de quoi faciliter la mise en place de exa-cel dans un hôpital qui souhaite apprendre la technique. À ce jour, plus de 60 patients ont été traités au niveau mondial par CRISPR-Cas9.
Une prouesse technologique
L'édition génomique ouvre une nouvelle ère dans le traitement des SCD et des TDT. L’effet de exa-cel est impressionnant et durable dans les études cliniques et confirmé en vie réelle. La faisabilité est démontrée en pratique quotidienne. Est-ce la solution miracle pour tous ?
Pour le Pr Colard, « ce sont des résultats impressionnants sur des patients sélectionnés pour des études, mais il ne faut pas oublier les réalités de la vraie vie, les c/i de nos patients (allo-immunisations, comorbidités … ), la complexité de la mise en œuvre, le coût important et un recul trop court (60 mois) pour juger de l’effet sur l’incidence des AVC, des atteintes cardio-pulmonaires, etc. Nous ne pourrons pas traiter tous nos patients, mais il n’en reste pas moins que pour ceux qui sont éligibles, Casgevy® est une prouesse technologique à laquelle il ne manque plus que le remboursement pour les patients de 12 ans et plus espéré pour le courant 2026 pour les deux hémoglobinopathies. »
Références
1. Locatelli F, et al. 30th Annual European Hematology Association 2025, Milan
2. Frangoul H, et al. 30th Annual European Hematology Association 2025. Milan
3. Frangoul H et al. ASH Annual Meeting 2025 Orlando, FL.