Galenusprijs ging vorig jaar naar ingenieuze behandeling hemoglobinopathie
Ernstige sikkelcelanemie en transfusiedependente ß-thalassemie kunnen worden behandeld met een ingenieuze technologie van ex-vivo bewerking met “moleculaire scharen” van genen van de hematopoëtische stamcellen van de patiënt. Het resultaat is exa-cel van de firma VERTEX, die daarvoor vorig jaar werd beloond met de Galenusprijs voor technologische innovatie.

Het biedt de gelegenheid deze twee zeldzame ziektes en de weg die al is afgelegd om de patiënten alsmaar beter te behandelen, in de schijnwerpers te plaatsen. Een succesverhaal verteld door prof. M. Colard (HUB Erasmusziekenhuis), prof. M. Algeri (Italië) en prof. Essa (Saudi-Arabië) op het 40ste congres van de Belgian Hematology Society.
Epidemiologie
Wereldwijd heeft 5,2% van de bevolking een of andere hemoglobinopathie. Volgens recente ramingen hebben tien miljoen mensen sikkelcelanemie (SCD) en worden jaarlijks 50.000 baby’s geboren met een transfusiedependente ß-thalassemie (TDT). TDT wordt vooral gezien in het Middellandse Zeegebied, het Midden-Oosten en Azië en sikkelcelanemie wordt vooral gezien in Afrika en India.
Prof. Colard: “Het zijn ernstige ziektes, die worden onderschat en die onvoldoende worden opgespoord. Tegelijk is het epidemiologische landschap aan het veranderen door migratie en een explosieve bevolkingsgroei in landen zoals Nigeria. België liep wat achterop. Sinds 2023 worden pasgeborenen systematisch gescreend, maar enkel in de Fédération Wallonie-Bruxelles."
De kliniek van SCD en TDT
De klinische verschijnselen van sikkelcelanemie zijn bloedarmoede, hevige pijn door vaso-occlusieve crisissen, chronische aantasting van de organen en een daling van de levenskwaliteit. Die patiënten leven minder lang. In Europa worden ze gemiddeld circa veertig jaar oud. (Levenslange) transfusies zijn moeilijk gezien de hoge frequentie van allo-immunisatie tegen de erytrocyten als gevolg van de ziekte zelf en de discordantie in bloedgroepen tussen Europese donoren en patiënten van Afrikaanse herkomst. 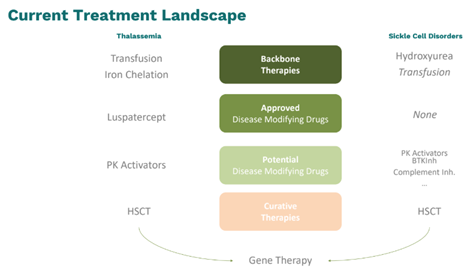
Sikkelcelanemie wordt behandeld met hydroxyureum. Dat verhoogt de hoeveelheid foetaal Hb, vermindert de sikkelvorming van de rode bloedcellen en voorkomt zo pijnlijke vaso-occlusieve crisissen.
Patiënten met een TDT vertonen een vaak ernstige chronische anemie met daardoor vermoeidheid en kortademigheid en extramedullaire hematopoëse. Ze moeten levenslang transfusies krijgen plus ijzerchelatoren om orgaanaantasting door ijzeroverbelasting tegen te gaan.
Hun levenskwaliteit is minder goed en gemiddeld worden die patiënten in Europa 50-55 jaar oud. Hydroxyureum (niet officieel erkende indicatie) en luspatercept verbeteren de erytropoëse. Veelbelovende geneesmiddelen in ontwikkeling zijn de pyruvaatkinaseactiverende middelen mitapivat en etavopivat.
De enige in opzet curatieve behandeling voor beide hemoglobinopathieën is een allotransplantatie van hematopoëtische stamcellen.
Waarom volstaat een allotransplantatie niet?
Allotransplantatie van hematopoëtische stamcellen geeft uitstekende resultaten zowel bij thalassemie als bij sikkelcelanemie. Het slaagpercentage ligt tegen 90%, maar verschilt volgens het land, de donor, de leeftijd en de comorbiditeit van de patiënt,... Maar er is een keerzijde aan de medaille.
Prof. Colard: “De overleving is al bij al goed, maar er zijn toch risico’s: risico’s in samenhang met de procedure, beenmergaplasie en transplantaat-versus-gastheerziekte (acuut in circa 20% van de gevallen en chronisch in 14% van de gevallen). De sterfte als gevolg van transplantatie is 6%, wat niet weinig is, zeker als er effectieve geneesmiddelen bestaan, die de overleving verbeteren. Niet iedere patiënt kan beschikken over een goede donor (HLA-compatibele broer of zus). Het slaagpercentage is lager boven de leeftijd van 15 jaar. De meeste getransplanteerde patiënten zijn echter ouder dan 15 jaar.”
Het komt er dus op neer dat je het risico neemt een ziekte op te lopen om een andere te genezen. Tegen die achtergrond wordt gezocht naar nieuwe behandelingen zoals pyruvaatkinaseactiverende middelen, brutontyrosinekinaseremmers en gentherapie (autotransplantatie van genetisch gewijzigde hematopoëtische stamcellen).
Gentherapie: van het lab naar het bed
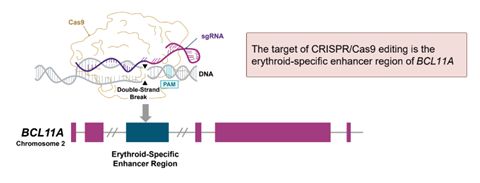 Door genetische engineering kan je gerichte genetische wijzigingen aanbrengen in onverschillig welk type cel. Dat gebeurt met specifieke nucleasen (moleculaire scharen) zoals de CRISPR-Cas9-technologie. Je isoleert de CD34+ hematopoëtische stamcellen van de patiënt, wijzigt die met CRISPR-Cas9 om het BCL11A-gen uit te schakelen teneinde de productie van foetale hemoglobine (HbF) weer aan te wakkeren en vervolgens dien je ze weer toe aan de patiënt.
Door genetische engineering kan je gerichte genetische wijzigingen aanbrengen in onverschillig welk type cel. Dat gebeurt met specifieke nucleasen (moleculaire scharen) zoals de CRISPR-Cas9-technologie. Je isoleert de CD34+ hematopoëtische stamcellen van de patiënt, wijzigt die met CRISPR-Cas9 om het BCL11A-gen uit te schakelen teneinde de productie van foetale hemoglobine (HbF) weer aan te wakkeren en vervolgens dien je ze weer toe aan de patiënt.
Het resultaat is exa-cel (exagamglogene autotemcel), waarbij geen virale vector wordt gebruikt zoals bij andere vormen van gentherapie. De resultaten zijn indrukwekkend. In de CLIMB THAL-111-studie(1) heeft 94,5% van de patiënten met een TDT (12-35 jaar) die werden behandeld met exa-cel, geen transfusies meer nodig gehad gedurende gemiddeld 40,5 maanden (mediane follow-up van 44,7 maanden).
In de CLIMB SCD-121-studie(2) heeft 91,1% van de patiënten met een sikkelcelanemie die exa-cel hadden gekregen, geen vaso-occlusieve crisissen meer vertoond gedurende gemiddeld 35 maanden (mediane follow-up van 39,8 maanden). De bijwerkingen zijn die van myeloablatieve conditionering en van autotransplantatie van hematopoëtische stamcellen. De resultaten waren nog beter bij kinderen (2-11 jaar) met een TDT.
In de CLIMB THAL-141-studie(3) heeft 100% van de patiënten tijdens de eerste 12 maanden geen transfusies meer nodig gehad en in de CLIMBSCD-151-studie(3) heeft 100% van de kinderen gedurende 12 maanden geen vaso-occlusieve crisissen meer vertoond. Exa-cel verbetert ook de levenskwaliteit van de patiënten te oordelen naar de aan de gezondheid gerelateerde levenskwaliteit.
En in het echte leven? De prevalentie van TDT en sikkelcelanemie is bijzonder hoog in Saudi-Arabië in vergelijking met de buurlanden in het Midden-Oosten. De populatie van Saudi-Arabië is bovendien erg homogeen. Er zijn beslisbomen ontwikkeld voor selectie van de patiënten, en het behandelingsprotocol is zeer gestandaardiseerd op alle niveaus: rekrutering van de patiënten, conditionering, voorbereiding van het transplantaat, manipulatie van het transplantaat en follow-up na verlaten van het ziekenhuis.
Alle demografische, klinische, laboratorium- en technische gegevens worden in een register bijgehouden om ziekenhuizen die de techniek willen leren, te helpen met de invoering ervan. Tot nog toe zijn wereldwijd meer dan 60 patiënten behandeld met CRISPR-Cas9.
Een technologisch hoogstandje
Genetische engineering opent een nieuw tijdperk in de behandeling van sikkelcelanemie en transfusiedependente ß-thalassemie. Zowel in klinische studies als in studies uitgevoerd in het reële leven is exa-cel op lange termijn bijzonder doeltreffend gebleken. De haalbaarheid ervan is aangetoond in de klinische praktijk. Is dat de mirakeloplossing voor iedereen?
Prof. Colard: “De resultaten bij de geselecteerde patiënten in klinische studies zijn ronduit indrukwekkend, maar je moet toch rekening houden met de realiteit van het echte leven, de contra-indicaties (allo-immunisatie, comorbiditeiten,…), de complexiteit van de behandeling en de hoge kosten. Bovendien is de follow-up nog te kort (60 maanden) om het effect op de incidentie van CVA, aantasting van het hart en de longen enz. te evalueren. We zullen niet alle patiënten kunnen behandelen, maar dat neemt niet weg dat Casgevy® een technologisch hoogstandje is voor de patiënten die ervoor in aanmerking komen. We hopen dat Casgevy® in 2026 zal worden terugbetaald voor patiënten van 12 jaar of ouder met sikkelcelanemie of thalassemie.
Referenties:
1. Locatelli F, et al. 30th Annual European Hematology Association 2025, Milaan
2. Frangoul H, et al. 30th Annual European Hematology Association 2025. Milaan
3. Frangoul H et al. ASH Annual Meeting 2025 Orlando, FL.